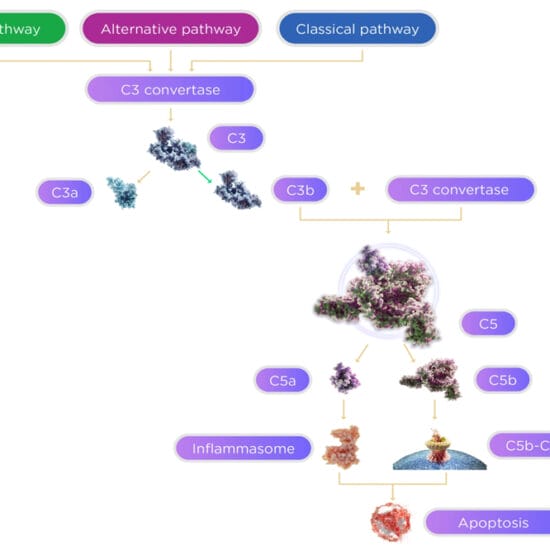
Giant cell arteritis is a rare disease with sight-threatening consequences unless detected and treated quickly and aggressively.
Giant cell arteritis (GCA) is a granulomatous vasculitis that affects medium-sized muscular arteries, particularly the cranial arterial branches of the aortic arch. Involvement of the ophthalmic artery and its tributaries can cause acute-onset vision loss. Risk factors include age (≥ 50 years), gender (female predominance) and being of Northern European/Caucasian descent.
Patients with GCA often present to their optometrist or ophthalmologist with anterior ischaemic optic neuropathy (AION) causing acute severe monocular vision loss

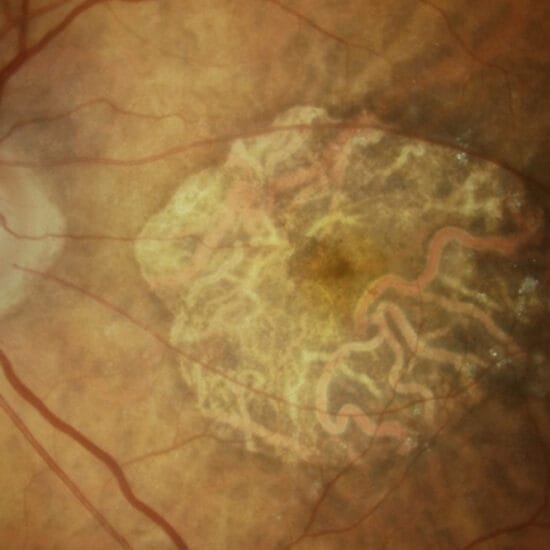
Figure 1a. Chalky white swelling of the right optic disc in a patient with giant cell arteritis related arteritic anterior ischaemic optic neuropathy.
The incidence of GCA is 15-20 per 100,00 in the over-50 age group, but rates are much lower outside of European population groups. GCA is rare in Asian and Black Caribbean/African populations. Women account for nearly 75% of all cases, with the lifetime risk being 1% compared to 0.5% in men.1
A genetic component to the development of GCA is supported by evidence of different prevalence in ethnic groups, familial aggregation and the strong association with certain human leucocyte antigen (HLA) regions.1 It remains unclear if previous infections have a role in the pathogenesis of GCA.2
Patients with GCA often present to their optometrist or ophthalmologist with anterior ischaemic optic neuropathy (AION) causing acute severe monocular vision loss, which can rapidly progress to become bilateral. Vision loss is often preceded by ischaemic symptoms of diplopia, amaurosis fugax or coloured positive transient visual obscurations (TVOs). These TVOs typically last minutes to hours and suggest transient choroidal ischaemia and hypoperfusion of the optic nerve head. Patients may also present with diplopia as the main presenting symptom, without any vision loss.

Figure 1b. OCT confirms disc oedema, with subtle changes on the left eye as well.
Systemic symptoms, including jaw claudication, headache and scalp tenderness, are important features to characterise in the clinical history. Jaw claudication refers to masseter muscle pain after a brief period of chewing. The presence of jaw claudication is one of the most helpful clinical symptoms, with the odds of a positive temporal artery biopsy (TAB) being nine times higher in those with jaw claudication.3 This should not be confused with temporomandibular joint dysfunction or dental disease.
Headache is typically new or different from previous headaches, severe and throbbing in quality, and either diffuse or localised to the temporal or occipital regions. The region over the temporal artery may be sensitive to touch. Patients with scalp tenderness often complain of pain when combing their hair, or an aversion to wearing hats.
Concurrent polymyalgia rheumatica in GCA is common, which manifests as morning stiffness and pain of the axial and large joints (neck, shoulders and hips). Prominent constitutional symptoms include malaise, weight loss and fever. Due to the wide-ranging phenotype, clinicians require a high index of suspicion for GCA. Occult GCA occurs in 20% of patients, where associated symptoms are absent.4
EXAMINATION

Figure 2. A seventy-year-old presented with count fingers right eye, 6/60 left eye vision. The main abnormality is the cotton wool spot on the left eye indicating ischaemia. ESR 95, CRP 110 and positive temporal artery biopsy.
Vision loss is often severe, to the magnitude of 6/60 or worse, particularly for GCA associated with arteritic AION. If vision loss is unilateral there will be a relative afferent pupillary defect. Colour vision will be reduced in eyes with an associated optic neuropathy. Eye movements and cranial nerve examination are important. Diplopia can be caused by single or multiple cranial neuropathies, or localised ischaemia of ophthalmic artery branches supplying the extraocular muscles.5,6
Fundus examination typically demonstrates diffusely ‘chalk-white’ optic disc oedema (Figure 1). Simultaneous occlusion of the central retinal artery and cilioretinal artery is a pathognomonic sign. Retinal cotton wool spots are also a sign of ischaemia which may be found in GCA (Figure 2). External signs of GCA are a thick, tender, non-compressible and non-pulsatile temporal artery.
Standard automated perimetry is useful in documenting visual field defects in GCA, particularly in the contralateral eye. NAION scotomas tend to occur in an altitudinal or arcuate pattern respecting the horizontal meridian, while central scotomas predominate in arteritic posterior ischemic optic neuropathy (PION).5
Optical coherence tomography (OCT) can be used to provide a baseline for the affected eye or show early oedema in the contralateral optic disc. However, a formal role for OCT in the assessment of GCA is not yet established. Non-specific vascular changes in the peripapillary and retinal vessels may be seen, but the significance requires further investigation.
Fluorescein angiography (FFA) is a useful adjunct for patients with vision loss, and a history suggestive of GCA, particularly when the TAB is negative or equivocal. Typically, the FFA will demonstrate delayed or patchy choroidal filling, and delayed central retinal arterial filling (Figure 3). This is particularly useful in distinguishing non-arteritic AION from arteritic forms.

Figure 3. Fundus fluorescein angiogram of the case from Figure 2, showing patchy delayed choroidal filling of the left eye.
DIAGNOSIS
There remain no validated diagnostic criteria for GCA. The American College of Rheumatology has classification criteria to distinguish GCA from other forms of vasculitis whereby patients should meet three out of five criteria: age ≥ 50 years, newonset localised headache, tender or reduced pulsatility of superficial temporal artery, ESR ≥ 50 and abnormal biopsy specimen. These should not be used in isolation and as a rule, all patients should undergo a TAB to confirm the diagnosis and assist in the long-term management.7 In experienced, high volume centres, specialised GCA protocol ultrasound and/or positron emission tomography/ computer tomography (PET/CT) studies may be used to confirm the diagnosis. While the European literature guidelines have suggested that a positive imaging study can be performed in preference to a TAB, this is yet to be endorsed by North American or Australian ophthalmic or rheumatology groups. Ultrasound can be useful, as it can assess not only the temporal artery but also the occipital, subclavian and axillary arteries which are not amenable to biopsy. If a temporal artery ultrasound is to be performed, the technician should assess all arteries before making a diagnosis.
As soon as the diagnosis is considered, the patient needs to be urgently referred to an ophthalmologist or directed to an Emergency Department for admission under neurology or rheumatology, with input from ophthalmology. High-dose steroid therapy should not be deferred for the TAB and should be commenced immediately, as it improves visual outcomes and prevents vision loss.8
Patients will typically have blood tests performed to measure markers of inflammation, in particular ESR, CRP and platelet count. A raised ESR and CRP is both sensitive and specific for GCA, though there will be approximately 10% of patients who have normal blood results.3

Figure 4. Thickened, tender and non-pulsatile temporal artery.
A TAB should be performed within 10 days, but up to four weeks from the time of starting prednisolone treatment. This is typically done in theatre by the ophthalmic team but can also be performed by a vascular or plastic surgeon. The biopsy needs to show inflammatory cells infiltrating the wall of the artery (transmural inflammation) to confirm a diagnosis of GCA. Other features reported may include fragmentation of the internal elastic lamina and thickening of the arterial wall, but these alone are not diagnostic.
GCA should not be a diagnostic consideration in anyone younger than 50 years of age. In any age group, rheumatological conditions associated with non-GCA arteritic AION that can mimic GCA include polyarteritis nodosa and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides.
MANAGEMENT
GCA is a sight- and life-threatening ophthalmic emergency and must be treated promptly. Delaying treatment can lead to disease progression to the fellow eye and irreversible vision loss in one third of patients within one day, one third in one week and one third in one month.9 Involvement of the carotid and/or vertebral circulation can cause transient ischaemic attacks or strokes. GCA has been notoriously challenging to manage. After many years, watching advances in other immune-mediated diseases, we are finally seeing progress in how we evaluate and treat this condition.

Figure 5. OCT confirmed increased signal intensity of inner retina and loss of definition of the nerve fibre layer margin, consistent with ischaemia on the right and superior segment swelling on the left.
High-dose steroid therapy (prednisolone) is often required for 18-24 months. Steroid treatment is often complicated by systemic side effects including diabetes, hypertension and osteoporosis. Ocular side effects include posterior subcapsular cataract and glaucoma. In one study of 120 GCA patients, 86% experienced at least one adverse event, the rate of events was correlated with increasing age and total cumulative dose.10
Patients who present with vision loss, are typically treated with 0.5-1gram of intravenous methylprednisolone for three to five days. However, there is no evidence that this reduces rates of vision loss. For those without vision loss or ocular ischaemia, 1mg/kg (to a maximum of 60mg/day) of oral prednisolone is used in the initial phase of treatment. There is no single accepted weaning regimen, though most clinicians will reduce the dose based on symptoms and inflammatory markers. One example of a tapering regimen published by the British Society of Rheumatology recommends; 40-60mg per day until symptoms and lab results are normal, reduce by 10mg every two weeks to 20mg, then 2.5mg every month to 10mg, and then 1mg every one to two months if there is no relapse.11
Over the past few years, a number of new medications have undergone clinical trials for GCA. One of these, a biological agent, tocilizumab (TCZ) was shown to have a steroid sparing effect (minimising the cumulative dose of prednisolone) and to prevent relapse. In 2017, tocilizumab (TCZ) became the first medication to be granted Food and Drug Administration approval specifically for the treatment of GCA. In Australia TCZ is now approved for use by rheumatologists, clinical immunologists and neurologists experienced in the management of GCA.
TCZ is an interleukin-6 (IL-6) receptor antagonist, which has been used in the treatment of rheumatoid arteritis and juvenile idiopathic arteritis for many years. There is evidence that IL-6 is important in the pathogenesis of GCA by promoting the systemic inflammatory response. Blocking IL-6 can abrogate this inflammatory cascade and control the symptoms, signs and inflammatory marker elevation inherent to this condition. However, it remains to be seen whether TCZ has a disease modifying, rather than suppressive, effect on the underlying pathophysiology.

Figure 6. Left eye inferior field loss.
The GiACTA trial was a randomised controlled trial, which enrolled 251 patients with newly diagnosed (47%) or relapsed (53%) GCA.12 The study compared a treatment strategy of subcutaneous TCZ with 26-week prednisolone taper versus placebo with 26-week prednisolone taper. It showed that TCZ had a steroid-sparing effect with sustained remission at 52 weeks in 56% of the weekly TCZ group compared to only 14% in the placebo group. The cumulative prednisolone dose was approximately halved in the TCZ group compared to the placebo group. When analysing the vision-related events in the patient groups, it appears that weekly TCZ is the best regimen. Longer term follow-up data from this study indicates that around half of patients experience relapse in the two years after TCZ is stopped.12 This means that all patients require careful long-term observation.
From a practical perspective, adding TCZ to the treatment regime in GCA has two main benefits; 1) it reduces the cumulate corticosteroid (prednisolone) dose requirement, and 2) it reduces the risk of relapse and ocular events while patients are taking the medication. It is thus most beneficial for patients who are at risk of steroid side effects (for example, those with obesity, diabetes, osteoporosis, or those with relapsing disease with prior use of steroids) and those with risk of progressive end-organ damage (limited visual reserve, aggressive aortitis or significant cerebrovascular or peripheral artery involvement).

Figure 7. PET scan showing increased FDG tracer uptake. (A) Bilateral vertebral arteries, (B) Bilateral maxillary arteries,
(C) Left superficial temporal artery, (D) Right facial artery.
Whether TCZ should be included in the routine care of all patients with GCA, at the time of diagnosis, is an evolving issue. Due to the high cost of the medication, the Pharmaceutical Benefits Scheme (PBS) currently funds a maximum 12-month supply of TCZ for patients with newly diagnosed or relapsed GCA. Further studies are required to determine the ideal duration of therapy and long-term outcomes of a 12-month course of therapy.
The main risk for the use of TCZ is infection, including reactivation of latent infections such as tuberculosis and Hepatitis B and C. That said, it does not confer a higher infection risk than a standard GCA corticosteroid regime. It is contraindicated in patients with a history of diverticulitis and gastrointestinal perforation. It may also cause liver toxicity and injection site reactions.
There are a number of other steroid sparing medications currently being trialled for GCA. These include Upadacitinib, Mavrilimumab, and subcutaneous methotrexate. It is important to note that oral methotrexate and leflunomide may also be used as steroid sparing agents in GCA, although the evidence for their effect is less convincing than that for TCZ.
REFERRAL TO RHEUMATOLOGY
Ophthalmologists have often been happy to treat and manage GCA with steroids, without physician input. Given the potential toxicities in the use of high-dose steroids, including osteoporosis, infection and steroid induced diabetes, formal rheumatological consultation was sometimes done. With the advent of new treatments, and in particular TCZ, we would now recommend early referral for the majority of patients. A rheumatology assessment in the era of biologic therapy will usually involve:
- Assessing for large vessel and aortic GCA involvement which is present in at least 50%of patients (GCA patients require long-term aortic surveillance due to risk of aneurysm),
- Glucocorticoid risk assessment,
- Screening for latent infection (especially TB, hepatitis) and other contraindications to biologics,
- Initiating Pneumocystis Jiroveci prophylaxis when appropriate, and
- Osteoporosis assessment. This will include documenting a history of fragility fractures, organising a bone mineral density scan and initiating bone resorptive therapy if required.
CASE ONE
A 65-year-old woman presented with horizontal double vision, worse for distance. The vision from each eye seemed unchanged. Over the previous week her husband noticed that she had switched to eating only soup. On further questioning she also mentioned that two weeks prior, her hairdresser had been a bit rough, pulling at her scalp and she no longer felt comfortable to wear her gold cap.
She had no past ocular or medical history of note and was not on any regular medication.
On examination, her vision was 6/6 in each eye. She had underaction of abduction of her left eye, with a distance esotropia of 25 prism dioptres, which reduced to six prism dioptres for near. Her temporal arteries were thickened and tender (Figure 4). The rest of the ocular and cranial nerve examination was normal.
A presumptive diagnosis of giant cell arteritis causing a sixth nerve palsy was made. She was given 60mg oral prednisolone. Her inflammatory markers showed ESR 110 and CRP of 54. Temporal artery biopsy confirmed GCA.
The woman’s symptoms resolved quickly with prednisolone. The inflammatory markers settled to normal levels over three weeks. Due to her good health she tolerated steroids well, with only some mild sleep disturbance and weight gain. Over the next 14 months her steroids were tapered, with no recurrence of symptoms nor change in her inflammatory markers. Two years later she was off all treatment and was doing well.
CASE TWO
An 88-year-old woman was admitted to hospital with progressive vision loss. She reported a three-month history of neck and shoulder stiffness and increasing fatigue. Over the previous two weeks she had also been experiencing temporal and frontal headaches. She denied jaw claudication. Her inflammatory markers were elevated with a CRP of 39mg/L and ESR of 89mm/h. She had a past history of glaucoma, ischaemic heart disease, chronic kidney disease and atrial fibrillation.
On examination, she had vision of light perception in the right eye and 6/9 left. She had a dense right relative afferent pupillary defect (RAPD). Fundoscopic examination revealed a pale swollen right optic disc on the right and superior disc swelling on the left, in keeping with arteritic anterior ischaemic optic neuropathy (AAION). OCT confirmed increased signal intensity of inner retina and loss of definition of the nerve fibre layer margin, consistent with ischaemia right and superior segment swelling on the left (Figure 5). Humphrey visual fields of the left eye confirmed an inferior altitudinal defect, consistent with superior disc infarction (Figure 6). She was immediately commenced on three days of intravenous methylprednisolone 500mg for presumed GCA.
She underwent a specialised GCA protocol PET/CT scan which confirmed the diagnosis of GCA with increased fluorodeoxyglucose (FDG) tracer uptake in the temporal, occipital, maxillary and vertebral arteries (Figure 7).
After commencing corticosteroid, she experienced worsening heart failure and steroid induced diabetes. She required diuretics and insulin.
Due to her aggressive ocular disease course, limited visual reserve and corticosteroid sideeffects, she was started on TCZ 162mg S/C weekly with a view to preserving vision and facilitate a more rapid wean of prednisolone.
At six-week follow-up she had no symptoms of GCA, her vision was stable and she was taking prednisolone 20mg and TCZ weekly. At the time of publishing, the patient was weaning prednisone over 26 weeks, in line with the GiACTA trial protocol. She will require close long-term follow-up to assess for relapse and a decision will be made at nine months as to whether any maintenance therapy is required in preparation for TCZ withdrawal at 12 months.
This article was commissioned by Roche Australia. The content is entirely independent and based on published literature and guidelines and the authors’ opinion. The views expressed do not necessarily reflect the views of Roche Australia.
To earn your CPD hours from this article visit mieducation.com/giant-cell-arteritis-detectionand- management
Associate Professor Clare Fraser specialises in neuroophthalmology and strabismus. She completed her ophthalmic training at Sydney Eye Hospital and neuroophthalmic training at Moorfields Eye Hospital and she also completed a research fellowship at Emory Eye Centre, Atlanta. A/Prof Fraser is an academic at the University of Sydney and Macquarie University, and consults at Liverpool Hospital, Sydney Eye Hospital and Macquarie University Hospital.
Professor Peter McCluskey is currently the Professor of Clinical Ophthalmology and Eye Health for the Faculty of Medicine and Health at The University of Sydney, and is the Director of the Save Sight Institute at Sydney Eye Hospital. He is an internationally recognised inflammatory eye disease specialist with more than 30 years’ experience treating patients with inflammatory eye disease.
Dr Anthony Sammel is a Sydney rheumatologist with subspecialty expertise in vasculitis. He is a staff specialist at Sydney/Sydney Eye Hospital, Prince of Wales Hospital and also sees patients at Randwick Specialist Centre. He works closely with ophthalmologists to assist with the assessment and management of patients with inflammatory and vasculitis-related eye disease. He runs a weekly dedicated vasculitis clinic and co-ordinates the Prince of Wales multi-specialty vasculitis clinical and research service. Dr Sammel also supports the Sydney/ Sydney Eye Hospital uveitis service. He completed his PhD in giant cell arteritis with a focus on the role of positron emission tomography (PET) scans to diagnose and monitor the disease.
Dr Jen Sandbach is a Sydney neuro-ophthalmologist and medical retina specialist, who provides the neuroophthalmology service at Prince of Wales Hospital. She also works privately at Annandale Eye Care and in Randwick.
References
- Lyons HS, Quick V, Sinclair AJ, Nagaraju S, Mollan SP. A new era for giant cell arteritis. Eye (Lond). 2020;34(6):1013- 1026. doi:10.1038/s41433-019-0608-7
- Gilden D, White T, Khmeleva N, Heintzman A, Choe A, Boyer PJ, et al. (2015) Prevalence and distribution of VZV in temporal arteries of patients with giant cell arteritis. Neurology. 84(19), 1948-55.
- Hayreh SS, Podhajsky PA, Raman R, Zimmerman B.Giant cell arteritis: validity and reliability of various diagnostic criteria. Am J Ophthalmol. 1997 Mar;123(3):285-96.
- Hayreh SS, Podhajsky PA, Zimmerman B. (1998) Occult giant cell arteritis: Ocular manifestations. American Journal of Ophthalmology. 125(4), 521-6.
- Haering M, Holbro A, Todorova MG, Aschwanden M, Kesten F, Berger CT, et al. (2014) Incidence and Prognostic Implications of Diplopia in Patients with Giant Cell Arteritis. The Journal of Rheumatology. 41(7), 1562-4.
- Smetana GW, Shmerling RH. (2002) Does this patient have temporal arteritis? JAMA. 287(1), 92-101.
- Murchison AP, Gilbert ME, Bilyk JR, Eagle Jr RC, Pueyo V, Sergott RC, et al. (2012) Validity of the American College of Rheumatology Criteria for the Diagnosis of Giant Cell Arteritis. American Journal of Ophthalmology. 154(4), 722-9.
- Hayreh SS, Biousse V. (2012) Treatment of acute visual loss in giant cell arteritis: should we prescribe high-dose intravenous steroids or just oral steroids? Journal of Neuroophthalmology. 32(3), 278.
- Danesh-Meyer H, Savino PJ, Gamble GG. (2005) Poor Prognosis of Visual Outcome after Visual Loss from Giant Cell Arteritis. Ophthalmology. 112(6), 1098-103.
10. Proven et al Glucocorticoid therapy in GCA: duration and adverse outcomes. Artheritis Rheum 2003; 49(5):703.)
- Dasgupta B, Borg FA, Hassan N, Alexander L, Barraclough K, Bourke B, Fulcher J, Hollywood J, Hutchings A, James P, Kyle V, Nott J, Power M, Samanta A, BSR and BHPR guidelines for the management of giant cell arteritis. Rheumatology (Oxford). 2010 Aug; 49(8):1594-7.
- Stone JH, Bao M, Han J, Aringer M, Blockmans D, Brouwer E, et al. (2019) OP0140 LONG-TERM OUTCOME OF TOCILIZUMAB FOR PATIENTS WITH GIANT CELL ARTERITIS: RESULTS FROM PART 2 OF THE GIACTA TRIAL. Ann Rheum Dis. 78(Suppl 2), 145-6.